![]()
![]()
![]()
![]()
![]()
Current Research
Understanding Plasmonic Catalysis from Accurate Excited-State Methods
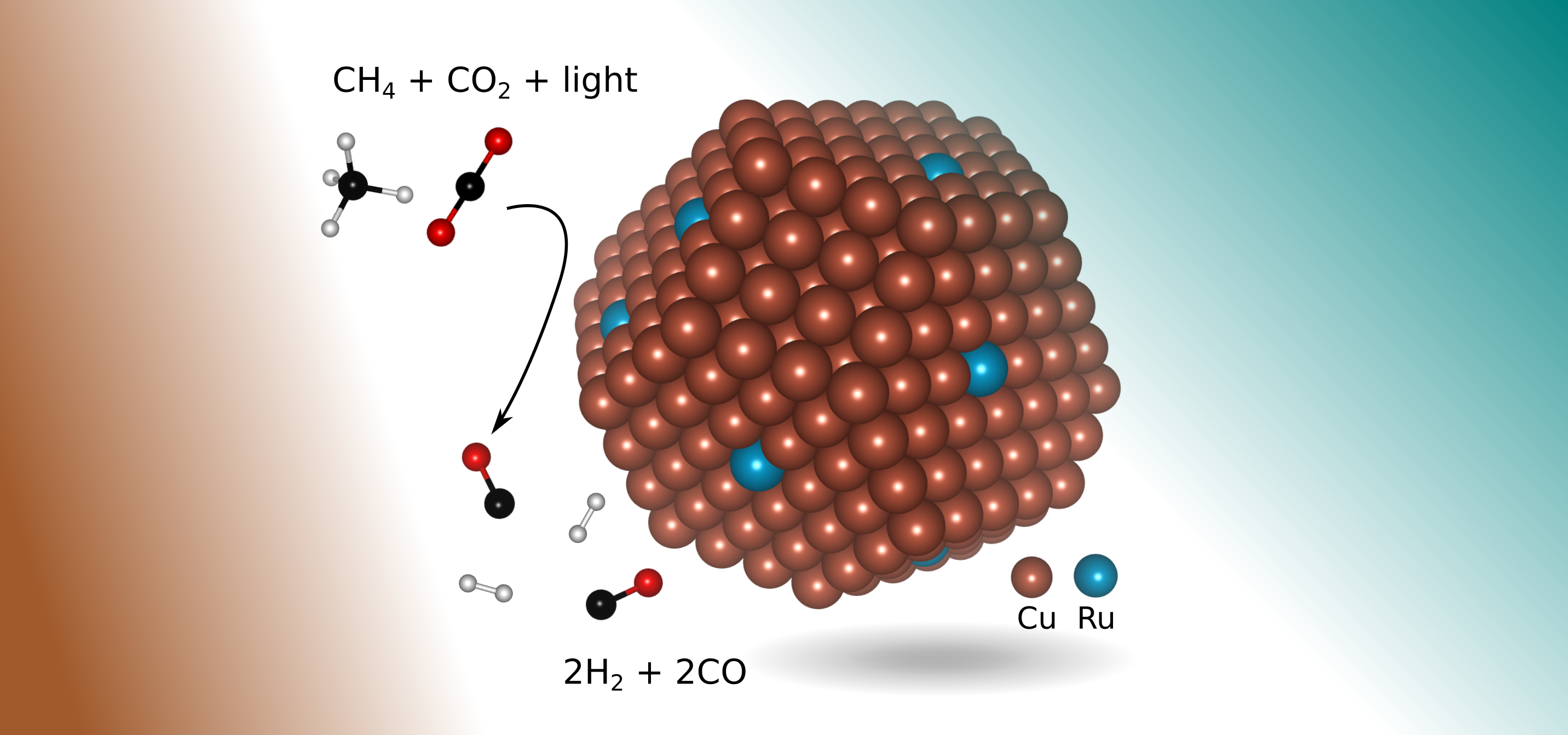
Summary. Our simulations furnished physical understanding and put forth reinterpretation of the effect of local surface plasmon resonances on heterogeneous catalysis. We addressed the kinetics of surface chemical reactions while considering electronically excited states. We concluded that plasmon resonances may lead to oscillating-dipole-induced local surface excitations making low-barrier, excited-state chemical reaction pathways available on the surface of plasmonic metals. This is in contrast to the energetic carrier injection and photothermal mechanism usually invoked to explain these phenomena.
Metallic nanoparticles (MNPs) with free-electron-like valence electrons, e.g., groups I and II metals, coinage metals (Cu, Ag, and Au), and Al, have enhanced ability to scatter and absorb light of wavelengths ranging from IR to UV. This size- and shape-controlled optical response of these MNPs is referred to as a local surface plasmon resonance (LSPR). LSPRs result in amplified surface and inter-particle electric fields, which then enhance light absorption of the molecules or other materials coupled to the MNPs, and/or generate hot carriers within the MNPs themselves. Plasmon-assisted catalysis on MNPs is an emerging technology that is potentially transformative for the field of heterogeneous catalysis because of the nanostructure’s ability to effectively lower reaction barriers using light. The exact mechanisms of action of the surface plasmon resonance that produces enhanced surface chemical reactivity on plasmonic catalysts are still in the early stages of investigation. We have shown and pioneered theoretical work that explores the possible role of resonance energy transfer and multiple electronic transitions on metallic surfaces in making possible otherwise high-barrier and difficult chemistries. These bodies of work contributed to understanding excited-state chemistry on surfaces of plasmonic MNPs using accurate quantum mechanical simulations. We used powerfully predictive embedded correlated wavefunction methods, within the density functional embedding theory to carry out these studies. Our studies on surface-doped plasmon-active metals with catalytically active elements also showed that surface dopants not only may lower reaction energy barriers, but also may act as energy funnels. Dopants achieve this by introducing dense frontier surface electronic states, effectively channeling collected light energy directly onto the chemical reaction.
Selected relevant primary-authored publications:
J. M. P. Martirez, J. L. Bao, and E. A. Carter, First-Principles Insights into Plasmon-Induced Catalysis. Annual Review of Physical Chemistry, 72, 99-119 (2021)
L. Zhou, J. M. P. Martirez, J. Finzel, C. Zhang, D. F. Swearer, S. Tian, H. Robatjazi, M. Lou, L. Dong, L. Henderson, P. Christopher, E. A. Carter, P. Nordlander, N. J. Halas, Light-driven methane dry reforming with single atomic site antenna-reactor plasmonic photocatalysts. Nature Energy, 5, 61-70 (2020)
J. M. P. Martirez, and E. A. Carter, Prediction of a Low-Temperature N2 Dissociation Catalyst Exploiting Near IR-to-Visible Light Nanoplasmonics. Sci. Adv., 3, eaao4710 (2017)
J. M. P. Martirez, and E. A. Carter, Excited-State N2 Dissociation Pathway on Fe-Functionalized Au. J. Am. Chem. Soc., 139, 4390-4398 (2017)
J. M. P. Martirez, and E. A. Carter, Thermodynamic Constraints in Using AuM (M= Fe, Co, Ni and Mo) Alloys as N2 Dissociation Catalysts: Functionalizing a Plasmon-Active Metal. ACS Nano 10, 2940-2949 (2016)
Chemical Insights into Oxygen Evolution Catalysis
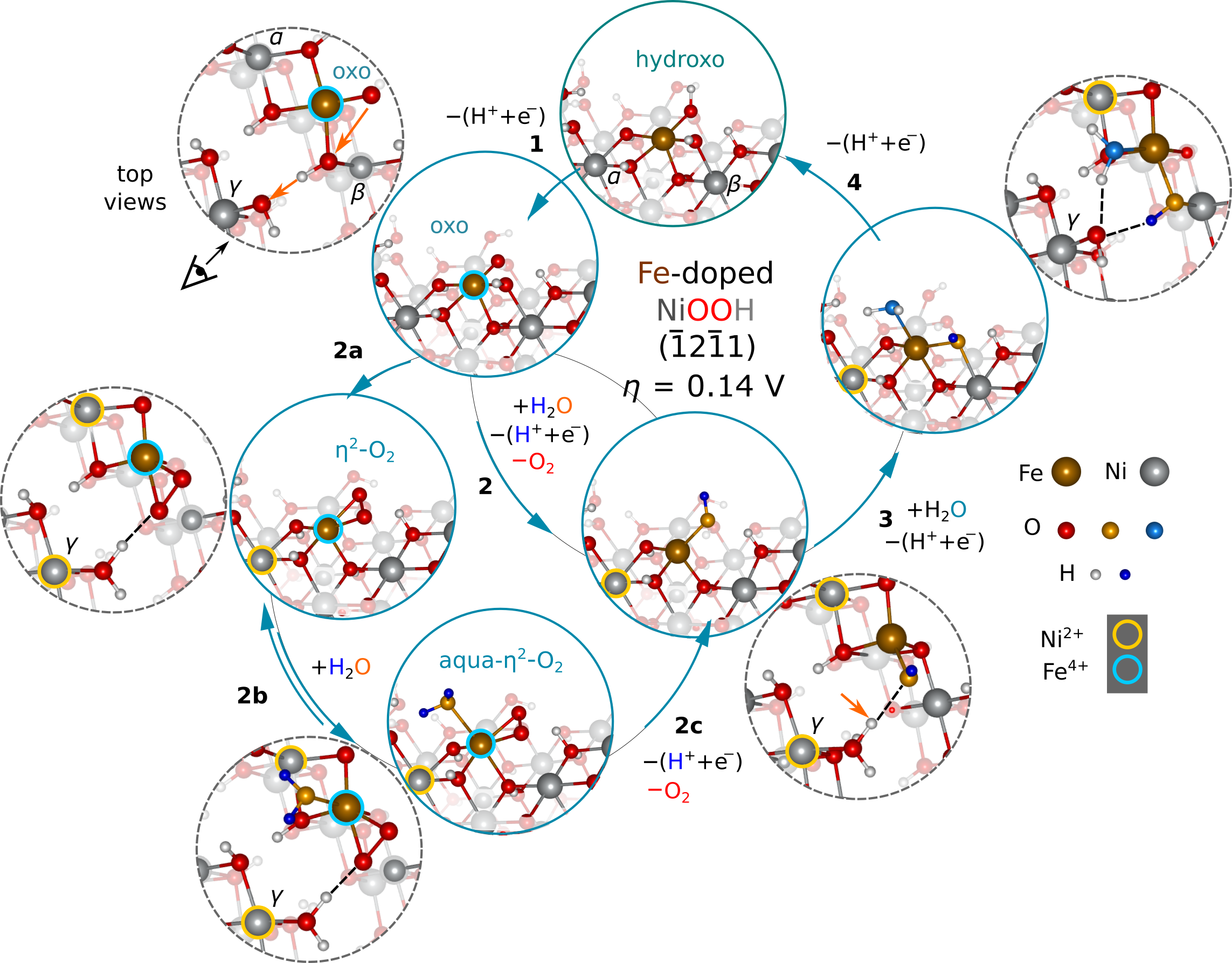
Summary. We identified key species and pathways in the electrochemical oxygen evolution on doped nickel-based oxyhydroxides. We were able to codify doping strategies to improve the aqueous electrocatalytic oxygen evolving activity of this material, which we based on two central mechanistic features of its efficient water oxidation. We thus were able to demonstrate a mechanism-oriented design principle in the development of oxygen evolution catalysts.
The electrochemical conversion of water into hydrogen and oxygen appears to be a viable path for on-site storage of the energy harnessed from intermittent renewable energy resources, which requires fast, high-capacity, and cheap methods of converting electrical energy into fuel. Optimization of the catalyst for the complex four-electron anodic oxygen evolution reaction is among the most pressing issues in the development of this technology. Fe-doped nickel oxyhydroxide (NiIIIOOH) has risen to the top as a cheap material with activities rivaling that of precious-metal-based catalysts. The exceptional performance of Fe-doped NiOOH has inspired studies to identify the central chemical and mechanistic features that govern the catalytic activity of this material. We evaluated the catalytic prowess of then unexplored molecular-like four-fold-lattice-oxygen-coordinated metal active sites, while using a more accurate hybrid density functional theory approximation, which is suitable for modeling redox reactions involving transition metal oxides. These distinguish our work from the current literature, and challenge widely implemented models. We codified the chemical origins of the site’s efficiency, while also unified prior experimental work that provided glimpses into the nature of the active site. We found dramatic differences between the O2 evolving performance of the Ni- and Fe-centered active sites, and unprecedented, near-quantitative predicted lower bounds for the electrochemical overpotentials. We showed that the O2 evolving activity of the Fe-doped β-NiOOH is a combination of the ability of surface Fe(III) to be stably oxidized as Fe(IV) with a terminal oxo, and the “dischargeable” nature of Ni(III) to Ni(II). We proposed and later investigated two doping strategies based on the above-mentioned two crucial mechanistic features. In our most recent papers, we showed that (1) a number of transition-metal dopants can be chemically modified when placed in a Ni(III) oxide environment, that many can be rendered ineffective as primary dopant. Fe(III) has near ideal redox synergism with Ni(III), unlike others. (2) Stronger oxidants compared to Ni(III), i.e., those that are found to the right of the periodic table, may promote O-O bond formation as secondary dopants. Our work highlights the utility of analyzing the redox behavior of transition metals in their molecular analogs in identifying important chemical features for efficient heterogeneous catalysts.
Selected relevant primary-authored publications:
J. M. P. Martirez, and E. A. Carter, Secondary transition-metal dopants for enhanced electrochemical O2 formation and desorption on Fe-doped β-NiOOH. ACS Energy Letters, 5, 962-967 (2020)
J. M. P. Martirez, and E. A. Carter, Noninnocent influence of host β-NiOOH redox activity on transition metal dopants’ efficacy as active sites in electrocatalytic water oxidation. ACS Catalysis, 10, 2720-2734 (2020)
J. M. P. Martirez, and E. A. Carter, Unraveling Oxygen Evolution on Iron-Doped β-Nickel Oxyhydroxide: The Key Role of Highly Active Molecular-like Sites. Journal of the American Chemical Society, 141, 693-705 (2019)
J. M. P. Martirez, and E. A. Carter, Effects of the Aqueous Environment on the Stability and Chemistry of β-NiOOH Surfaces. Chem. Mater., 30, 5205-5219 (2018)
A Simple Embedding Method for Covalent and Ionic Materials
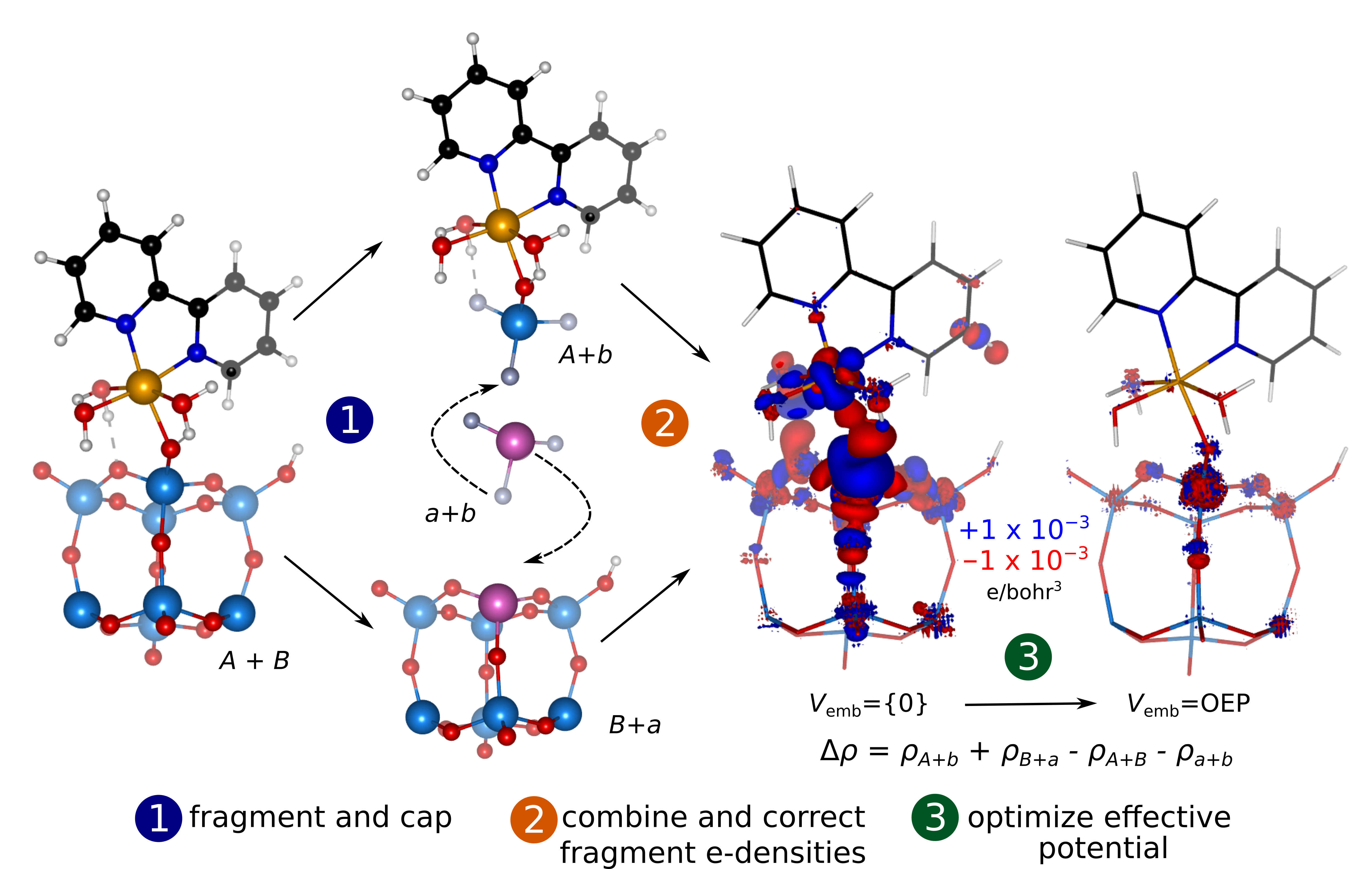
Summary. Density functional embedding theory (DFET) enables one to perform accurate but computationally expensive quantum mechanical calculations on chemical systems through fragmentation. We introduced an extension of DFET to covalent and ionic compounds, where the original DFET method had been shown to be inadequate. The goal is to perform accurate embedded correlated wavefunction calculations and evaluate the absorption and emission behavior of, for example, hybrid organic-inorganic systems. In such systems, the fundamental electronic transitions usually are localized between the organic chromophore and the nearest transition metal cation of the inorganic component of the semiconductor, making such systems suitable for the localized description of excited states (excitons).
Fully ab initio and systematically improvable correlated wavefunction (CW) methods are ideal for obtaining an accurate quantum mechanical description of chemical systems. However, they are computationally demanding and scale poorly with system size. Divide-and-conquer approaches in which large systems are partitioned into smaller fragments and where the fragment interactions are described using a low level of theory while the important fragment is described within a preferred higher level of theory, offer ways to incorporate CW in chemical simulations of large systems, e.g., biomolecules, surfaces, large inorganic clusters, bulk crystals, etc. We proposed a partitioning protocol that involves the use of capping atoms to saturate severed covalent bonds at fragment interfaces, which arises when a covalent system is partitioned into subsystems. We then use DFET to describe subsystem interactions. In our proposed protocol, the capping groups in each fragment and the auxiliary fragment (a separate system composed of only the combined capping groups) function as ad hoc regional correction to the potential and electron density, respectively. The method depends only on the capped-subsystems’ and auxiliary-fragment’s densities, which then avoids the use of orbital projectors to partition the total electron density. This makes its implementation straightforward in planewave-DFT codes. We demonstrated the utility and advantages of our capped-DFET and ensuing embedded capped-CW method in predicting reaction energy surfaces for two systems, namely, organic molecules and ionic metal oxides. We also demonstrated that with capped-DFET and embedded capped-CW theory, a plethora of metal-to-ligand charge-transfer excited properties of an organic-inorganic hybrid system can be simulated accurately at a reasonable computational cost. We envision the use of the capped-DFET method to probe similar systems used in optical devices involving semiconductors combined with a range of organic molecules.
Selected relevant primary-authored publications:
J. M. P. Martirez and E. A. Carter, Metal-to-ligand-charge-transfer spectrum of a Ru-bipyridine-sensitized TiO2 cluster from embedded multi-configurational excited-state theory, Journal of Physical Chemistry A, 125, 4998-5013 (2021)
J. M. P. Martirez and E. A. Carter, Projector-free capped-fragment scheme within density functional embedding theory for covalent and ionic compounds, Journal of Chemical Theory and Computation, 7, 4105-4121 (2021)